Amyotrophic Lateral Sclerosis or ALS, more commonly known as Lou Gehrig’s disease, is a terminal and progressive motor neuron disease. ALS specifically targets and kills the motor neurons responsible for controlling the vast majority of skeletal muscles in the human body, which eventually leads to respiratory failure and death.


A positive diagnosis of ALS is based primarily on a patient’s symptomatology as no test can currently provide a more conclusive assessment.
Unfortunately, there are many diseases whose symptoms resemble those observed in patients with ALS. Therefore, diseases such as cervical osteoarthritis, cervical hernias that compress the spinal cord, heavy metal poisoning, and some infectious diseases such as Lyme disease or syphilis, can delay a correct diagnosis of ALS.
As such, when ALS is suspected, it is common practice to rule out other diseases through a variety of tests including but not limited to lumbar punctures, MRIs, and electromyographic studies. In some cases, it might be necessary to perform a biopsy of muscle tissue in order to assuage any remaining doubts.
Often, the earliest symptoms of ALS are ignored or outright dismissed. Therefore, In an attempt to better understand this dreaded disease, we provide you with a list of the most common symptoms.
1. Fatigue


ALS is a syndrome with a varied evolution, and no two patients experience the same progression of symptoms. However, one of the earliest and most common signs of ALS is fatigue.
Fatigue in Amyotrophic Lateral Sclerosis can be best described as a sensation of persistent debility or exhaustion. It is one of the first symptoms to develop, and can affect patients for hours, days, or become chronic and exist for months at a time. Fatigue is extremely detrimental to a patient’s quality of life because it can severely hinder their ability to work or participate in family or social life.
Since fatigue is not an obvious symptom, those affected by ALS often feel misunderstood by people around them who do not perceive or understand the impact it can have on the life of the person who suffers it.
2. Loss of Strength

ALS belongs to, and is perhaps the most common example of, a group of neurological disorders known as Motor Neuron Diseases. These diseases affect the body by causing the death of millions of neurons found in the motor cortex of the brain as well as the spinal cord. These nerve cells are directly responsible for the regulation and control of skeletal muscle function.
As such, patients who have ALS eventually lose the ability to control all voluntary movement. During the progression of the disease, which typically lasts for several years, patients will experience a cumulative loss of muscle strength.
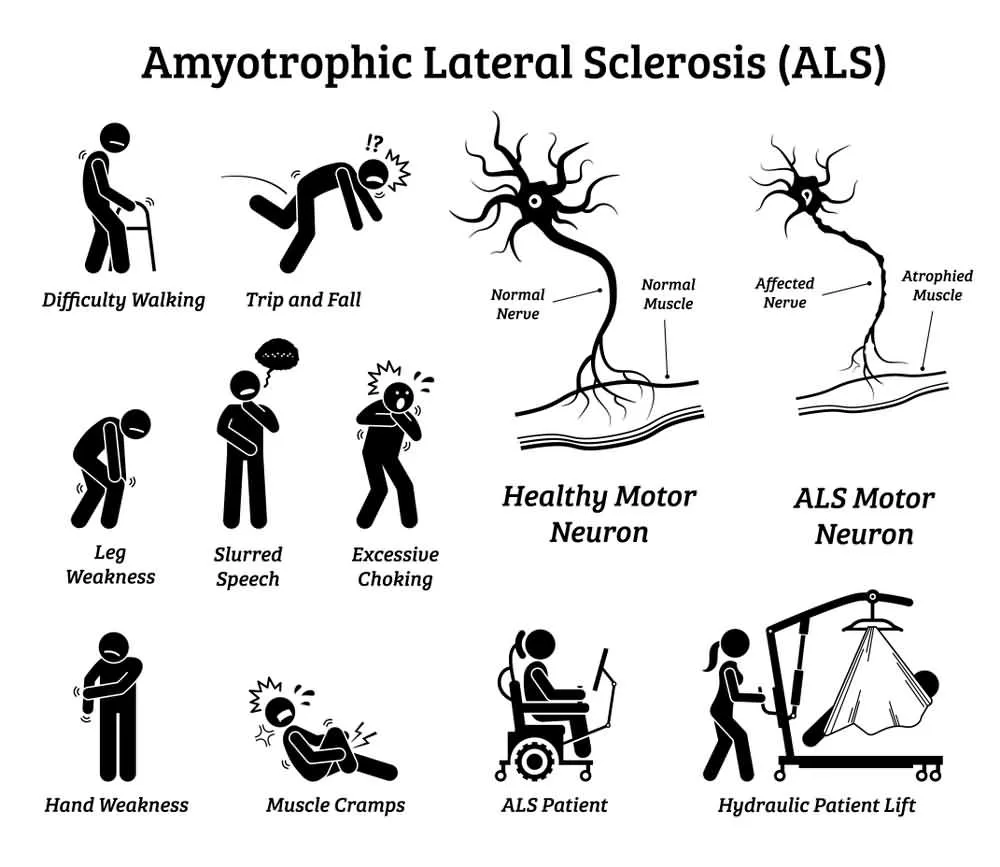
In most cases, the first muscles affected by the disease are those of the arms and legs which results in patients experiencing awkwardness when walking or moving about, an increased propensity for stumbling or tripping, and difficulty performing everyday tasks that require dexterity, such as tying shoelaces or typing on a keyboard.
3. Muscular Atrophy
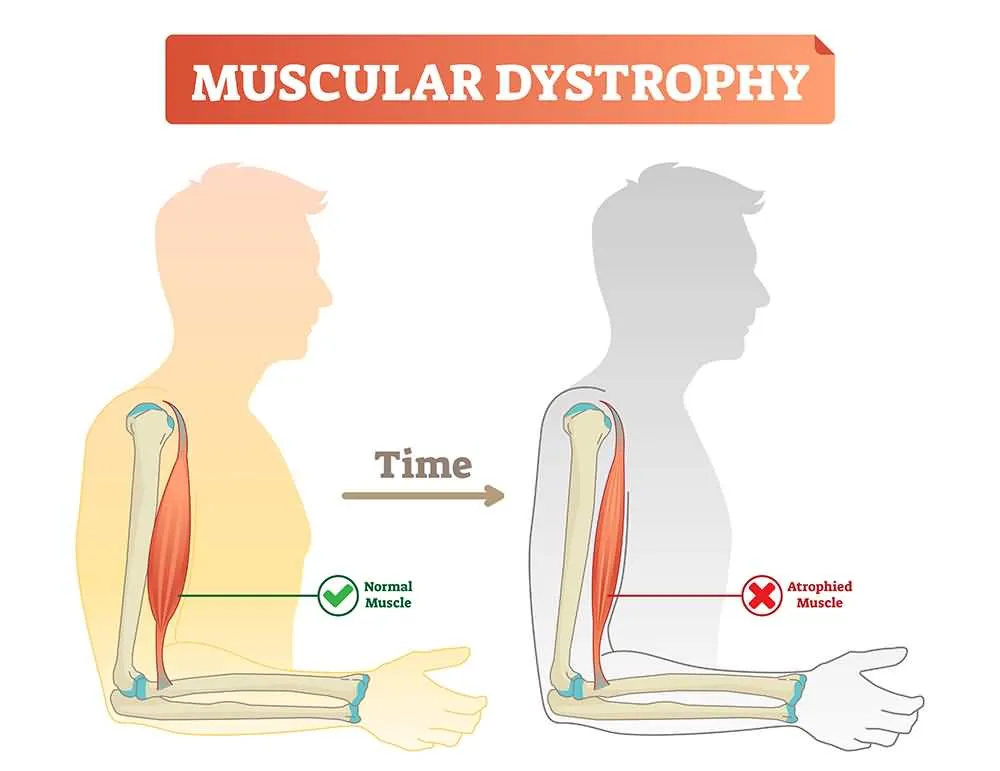

Muscular atrophy is a disorder that involves the loss or diminishing of the body’s skeletal muscle mass. It is generally caused by an abnormal imbalance between protein synthesis and its subsequent degradation. In the specific case of ALS, it occurs due to a dramatic reduction in the connection between nerves and muscle fibers caused by the death of motor neurons.
Because muscular atrophy directly affects the nerve cells of skeletal muscles, it often culminates in partial or total paralysis. This disorder contributes to the further loss of muscle strength, which in turn contributes to exacerbating atrophy. Progressively the muscles wear out, and the patient has more and more difficult to perform activities such as walking.
Muscle atrophy can be best characterized by the various symptoms that develop as the atrophy worsens. The most frequent signs of atrophy are a decrease in muscle mass of the arms and legs, a pervasive sensation of weakness in the extremities, and difficulty performing basic everyday tasks.
4. Fasciculations
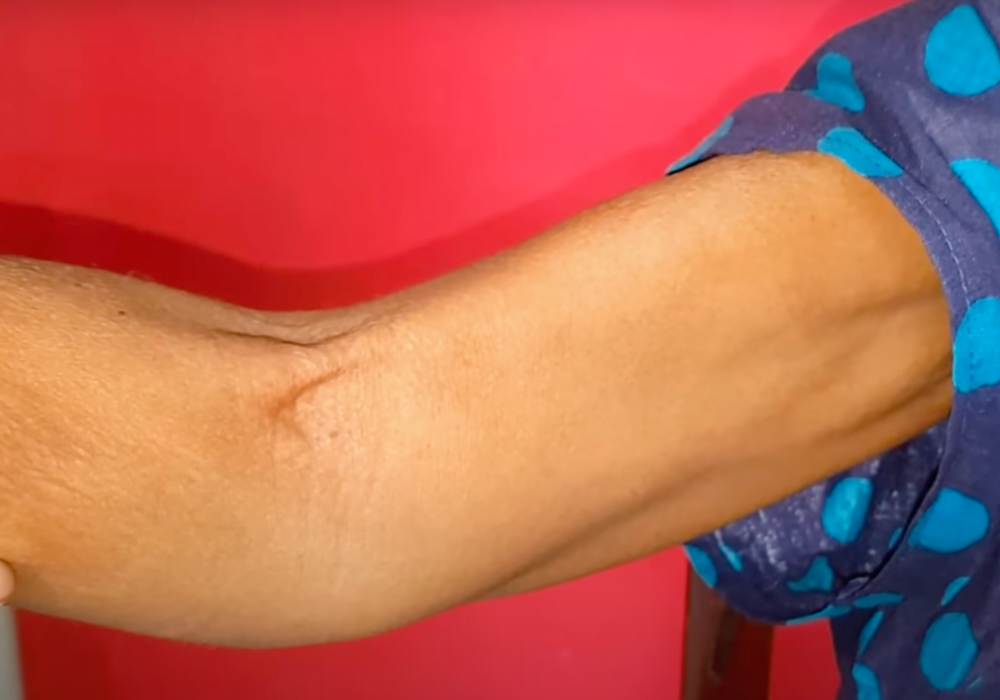

Fasciculations are slight and involuntary muscular contractions that occur underneath the skin, but that do not produce any observable limb movement. Fasciculations are visible to the naked eye and are sometimes described as looking like small worms are moving within the muscle. These contractions occur because of spontaneous nerve discharges that fire within clumps of skeletal muscle fibers.
Fasciculations are typically benign, but sometimes they can be caused by neurological diseases, such as Amyotrophic Lateral Sclerosis. In the specific case of ALS, the fasciculations occur due to damage present in the lower motor neurons. They can be considered an early warning sign of the possible onset of ALS.
5. Cramps
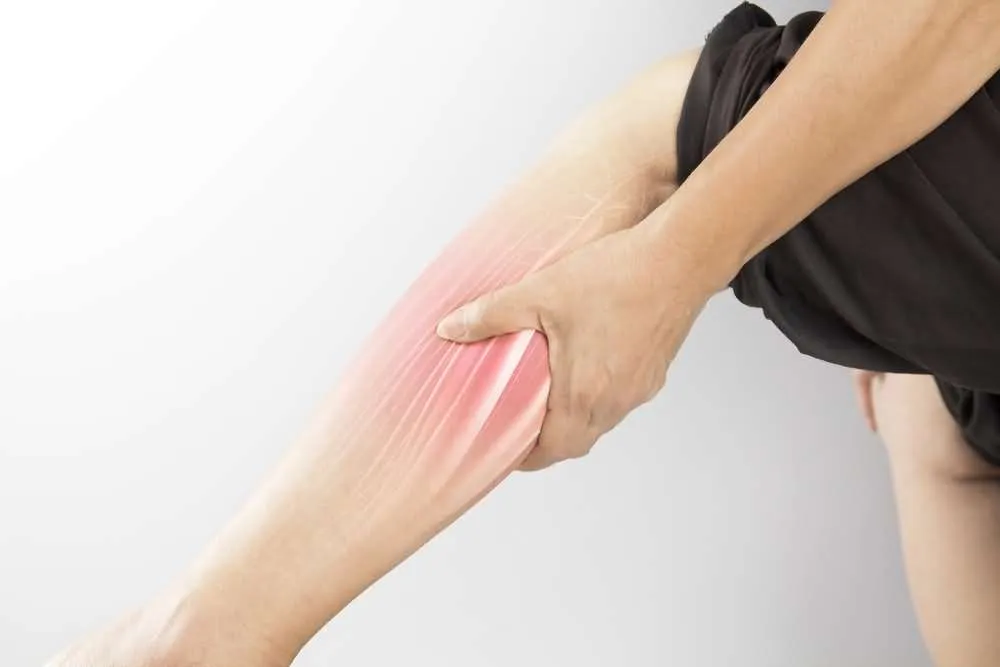
Muscle cramps are highly common in patients who have ALS, and their incidence increases as the disease progresses. These sustained involuntary contractions of the muscles are typically accompanied by palpable contractures, can last anywhere from 30 to 45 seconds, and tend to be extremely painful.
As a direct consequence of frequent muscle cramps, spasticity may develop if specific muscle groups are unable to relax sufficiently. Spasticity may hinder certain movements in which antagonistic muscle groups participate.
6. Dysphagia


Dysphagia is a difficulty or inability to swallow. In some cases of ALS, the first manifestations of the disease appear in the bulbar innervation muscles which severely impairs the patient’s ability to speak and swallow. Even in cases where the onset of ALS occurs along the spinal cord, at some point during the evolution of the disease, the bulbar dysfunction develops.
Dysphagia is a serious side effect as it can pose a clear danger to the patient’s life. In healthy subjects, delicate coordination between respiration and swallowing prevents food from entering the airways. However, ALS patients suffering from dysphagia loose this preventive function and can easily aspirate food into the respiratory tract.
7. Dysarthria


Due to the fact that Amyotrophic Lateral Sclerosis is a progressive disease of the cells of the anterior horn of the medulla, of the motor nuclei of the cranial nerve and of the corticospinal and corticobulbar pathways, and that it damages both upper and lower motor neurons, many patients develop dysarthria, or a difficulty to pronounce words.
The speech of ALS patients is characterized by presenting varying degrees of spasticity, slowness, excessive pauses, and a generalized difficulty pronouncing some phonemes. Their voice tends to be monotonous and with diminished tonal extension. The strength and rhythm of lip movements are reduced, and paralysis or velar paresis can develop. The tongue also becomes hypertrophic and prone to experiencing fasciculations.
8. Dyspnea

Typically, patients with ALS experience a progressive decrease in vital lung capacity due to a significant weakening of the chest muscles and diaphragm. Virtually all patients with ALS will ultimately experience shortness of breath when walking, or during rest, difficulty or inability to lie in bed and maintain respiratory capacity, a weakened cough reflex, and a severe inability to clear their throat and pulmonary secretions.
The development of dyspnea can be considered a turning point in the disease’s evolution since the vast majority of ALS patients die of respiratory failure.
9. Emotional Incontinence


Also known as the Pseudobulbar affect, emotional incontinence refers to a loss of the cortical control of emotions that is characterized by episodes of disproportionate and uncontrollable crying or laughing. Often, these episodes are mood incongruent, meaning patients will laugh during sad situations and vice versa.
Emotional incontinence outbursts typically occur several times a day with each episode lasting several minutes at a time. This symptom, while not necessarily clinically significant, does generate a great deal of social anxiety in most patients because it can adversely influence their ability to relate to others. It is important to emphasize that ALS patients retain full cognitive control and that this condition is uncontrollable and any outbursts are entirely involuntary.
10. Weight Loss

As a degenerative neurological disease, ALS has a significant impact on the nutritional status of patients. Therefore, patients with ALS, especially if presenting dysphagia, have an elevated risk of malnutrition, which in turn, exacerbates the loss of muscle strength and muscle mass that is so prevalent during the evolution of the disease.
The principal mechanisms that are conducive to weight loss in patients with ALS are a reduced caloric intake due to dysphagia and weakness of the arms, which make the ability to ingest food a challenging endeavor. Furthermore, patients experience a rise in energy expenditure due to an elevated metabolic rate as a result of increased respiratory effort.
Weight loss stands as one of the most important independent predictors of the quality of life and survival prognosis of patients affected by ALS as it is directly linked with higher mortality.
11. Pneumonia
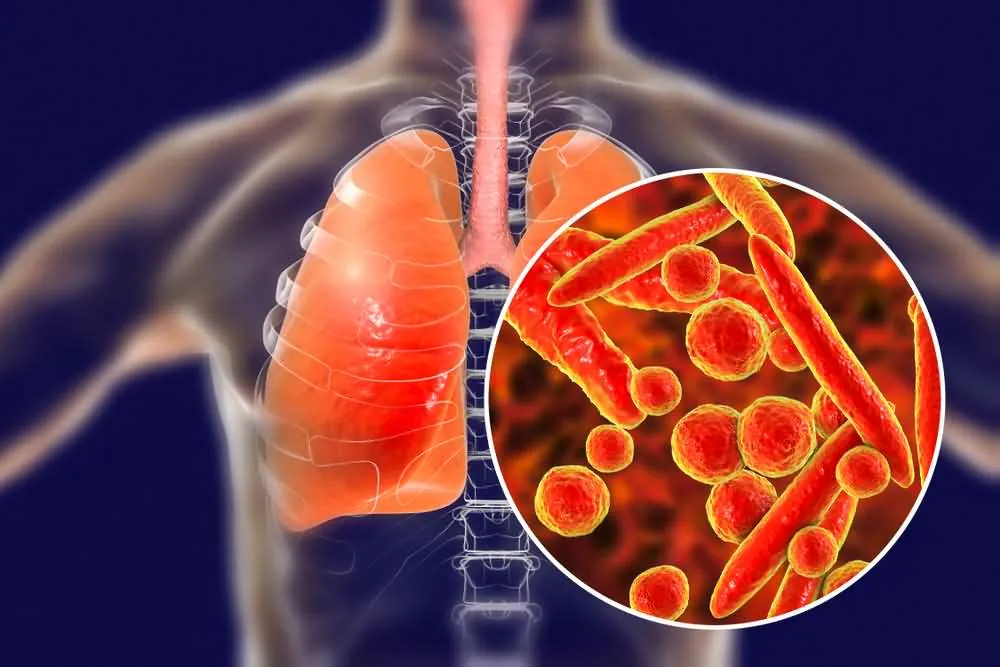
Pneumonia is not a symptom of ALS in and of itself; however, it stands as one of the most common and deadly complications of the disease.
Pneumonia is a respiratory infection that causes severe inflammation of the pulmonary alveoli, usually caused by bacteria or viruses. It is characterized by general malaise, a fever that is frequently preceded by chills, severe shortness of breath, productive cough, and chest pain. Pneumonia cough is commonly accompanied by yellowish sputum that may present traces of blood. If not treated effectively, pneumonia presents a risk of death.
In the case of patients with ALS, the evolution of pneumonia is complicated by the respiratory deficit already present as a consequence of the disease, the weakness of the chest muscles involved in breathing, and the difficulty, or outright inability, to expel the phlegm and mucus generated by the infection.
12. Cerebral Hypoxia


Cerebral Hypoxia represents another dangerous complication of Amyotrophic Lateral Sclerosis due to the diminished respiratory capacity of patients, especially at night. Cerebral Hypoxia is a condition in which there is a significant reduction in the supply of oxygen to the brain even if there is adequate blood flow.
Symptoms of cerebral Hypoxia include difficulty concentrating and paying attention, memory loss, and diminished coordination. Brain cells are extremely sensitive to oxygen deprivation and can begin to die within a few minutes of even a moderate decrease in the brain’s oxygen supply.
As such, much of the cognitive dysfunction experienced by ALS patients is due to cerebral Hypoxia.
13. Pain

Although pain is typically not a relevant symptom in the early stages of ALS, it is quite common in the later stages of the disease, with patients reporting discomfort in the lumbar region, as well as along the shoulders, neck, and legs.
Pain in ALS is thought to be caused by the progressive chronic stress suffered by the bones and joints of the body due to the muscular atrophy that characterizes the disease. Muscular contractures, cramps, and spasticity, as well as joint stiffness, only exacerbate the issue.
Furthermore, the progressive immobility experienced by patients in the later stages of ALS is conducive to the formation of painful ulcerative lesions due to ischemia.
» Conclusion

Amyotrophic Lateral Sclerosis affects approximately 5 out of every 100,000 people around the world, and in the United States alone about 5,000 new cases are diagnosed every year. ALS affects all races, ethnicities, and social groups equally.
Generally, the disease begins in the hands or feet, but as the condition progresses and more muscles are affected, it begins to impact the entire body. Thankfully, the vast majority of patients with ALS do not suffer any form of cognitive impairment or loss of sphincter control.
The exact causes behind the appearance of Amyotrophic Lateral Sclerosis are unknown. Currently, most research suggests that genetic and environmental factors play a significant role in its development. For example, it is suspected that about one-tenth of all ALS cases are generated through a genetic defect. Nevertheless, most affected patients possess no immediate or apparent causal factors.
Each year, as research advances and the scientific community learns more about the disease, the outlook of patients improves. Currently, a significant portion of diagnosed cases can live up to 10 years after the appearance of the disease thanks to significant improvements in clinical management and patient care.
References
Rowland, Lewis P., and Neil A. Shneider. “Amyotrophic lateral sclerosis.” New England Journal of Medicine 344.22 (2001): 1688-1700.
Kiernan, Matthew C., et al. “Amyotrophic lateral sclerosis.” The lancet 377.9769 (2011): 942-955.
Mitchell, J. Douglas, and Gian Domenico Borasio. “Amyotrophic lateral sclerosis.” The lancet 369.9578 (2007): 2031-2041.
Caroscio, James T., et al. “Amyotrophic lateral sclerosis: its natural history.” Neurologic clinics 5.1 (1987): 1-8.
Kühnlein, Peter, et al. “Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis.” Nature Reviews Neurology 4.7 (2008): 366.
David, P., et al. “Clinical features of amyotrophic lateral sclerosis.” The Italian Journal of Neurological Sciences 2.2 (1981): 111-117.