

Abnormal bleeding was not as uncommon in Germany, England, Spain, and Russia back in the 19th century. Hemophilia is one of the leading causes, and it affected many royal families to the point of being called “the royal disease.” For example, Queen Victoria of England was a carrier of a hemophilia type known as Hemophilia B. She had nine children, and 3 of them suffered from severe bleeding episodes due to hemophilia.
But what is hemophilia? If Queen Victoria had Hemophilia B subtype, what other types are there? How is it treated or managed? We’re covering all of these aspects of hemophilia and more in this article.
What is hemophilia?


Hemophilia refers to a genetic disorder associated with pathologic or abnormal bleeding. Blood clotting does not work as it should, and the patient often has spontaneous episodes of bleeding and slow healing with continuous bleeding from wounds and injuries. For this reason, patients with hemophilia need to be evaluated and carefully managed if they ever need to undergo a surgical procedure.
This disease shows up in one in 5,000 males. According to statistics gathered until 2018, up to 33,000 males were diagnosed with hemophilia in the United States. As we’re covering in this article, there are two types, too, and Hemophilia A is the most common.
In the blood, several proteins contribute to clotting. They are called clotting factors, and we have several of them. People with hemophilia have a deficiency of specific clotting factors, usually factor VIII or factor IX. Depending on clotting factor levels, hemophilia can be mild, moderate, or very severe, with or without complications and long-term consequences.
The disease usually starts early in life, but it may also develop later in older adults or middle-aged people. Sometimes, younger women develop hemophilia during pregnancy or after giving birth, but these cases are usually resolved after appropriately treating the condition.
» Now, let’s discuss types of hemophilia.
Types of hemophilia


The primary forms of hemophilia include Hemophilia A and Hemophilia B. However, we can also include acquired hemophilia as a separate entity:
› Hemophilia A
It is caused by defective or insufficient production of Factor VIII. This clotting factor is commonly produced in the liver’s vascular endothelium. Thus, liver transplantation usually corrects the problem in individuals with hemophilia A. This clotting factor works in association with the von Willebrand factor, which stabilizes and protects factor VIII from degradation.
The affected gene in hemophilia A is located on the Xq28 region of the chromosome X, which is found in the long arm, and it is known as F8C. It’s a very large gene, encoding for a protein with 2332 amino acids. In 40% of cases, the genetic defect comes from an inversion of the gene, but the remaining 60% of cases are caused by a point mutation, an insertion, or a deletion of the gene. The result is a dysfunctional or deficient FVIII, an abnormal cascade of coagulation, and excessive bleeding. The most common manifestation is joint bleeding, and intracranial hemorrhage usually appears in young patients before turning 18 years old.
› Hemophilia B
It is caused by defective or insufficient production of Factor IX. This problem disrupts the coagulation cascade, and excessive hemorrhage, usually located in the joints, and in the central nervous system, the muscles, pulmonary system, gastrointestinal tract, and other organs. It is less frequent as compared to hemophilia A, but shares many similarities. Factor IX is also synthesized in the hepatocyte and has a half-life of 18 to 24 hours. Like FVIII, FIX is also inactive in the blood, and when activated, they work together to activate factor X, which converts fibrinogen into fibrin and stops bleeding. Factor IX gene is located in the region Xq27, which is also found on the long arm of chromosome X. This gene is also known as F9, and encodes for a shorter protein (415 amino acids) as compared to FVIII. Deletions and point mutations are the most common cause of the disease. The clinical manifestations are basically the same as hemophilia A.
› Acquired hemophilia
It is caused by the formation of antibodies against clotting factors. It may cause a type of hemophilia A or hemophilia B, which appears later in life instead of being born with it. Around 30% of patients with severe cases of hemophilia A and 3-5% of patients with hemophilia B develop antibodies against clotting factor VIII, which may or may not be affected by a genetic mutation. Such inhibitors are immunoglobulin G subclass 4. When they are in contact with clotting factors, they neutralize their clotting effects in the coagulation cascade. This condition can be usually found in older adults (50 years old and later on). It is often associated with a collagen vascular disease or an adverse drug reaction against penicillin or another drug. A patient with very high levels of antibodies against FVIII should be evaluated for lymphoproliferative malignancies and other cancer types.
Hemophilia causes


The cause of hemophilia is a mutation to one or more genes that encodes clotting factors. These genes are translated into proteins, and these proteins are essential for coagulation. Having a defective gene or having it depleted leads to a faulty or absent protein and subsequent coagulation problems. These clotting genes can be found in the X chromosome found in males (XY) and females (XX). Thus, this is a sex-linked disease.
Hemophilia is a recessive disorder, which means that you need to have both X chromosomes taken to suffer the disease. Unluckily for males, they only have one of them, and they will have hemophilia whenever their X chromosome copy has the mutation. In women, having two X chromosomes allow them to be carriers of the disease without any symptom. The other copy of chromosome X does the job when it is intact, and they will only have hemophilia when both X chromosomes are affected by the disease. That’s why men are more likely to have hemophilia, and women would only suffer from the disease if they are born from men with hemophilia and carrier women who may or may not have the same condition.
Females can be carriers of hemophilia without having any symptoms. They are often unaware of their defective gene until they pass the mutated chromosome on to their offspring.
Hemophilia genes run in families, but sometimes they are not entirely evident. There are families with no prior history of hemophilia cases because they mainly have female carriers, and males in the family are not affected. But then, the first relative with hemophilia shows up, revealing the inheritance of the mutation in the X chromosome.
In some cases, hemophilia can be further complicated by the development of inhibitory antibodies to clotting factors. When this is the case, lacking the clotting factor is not the only problem, and treatment becomes more difficult and expensive.
» Continue reading to know more about hemophilia symptoms
Hemophilia symptoms
1. Slow healing
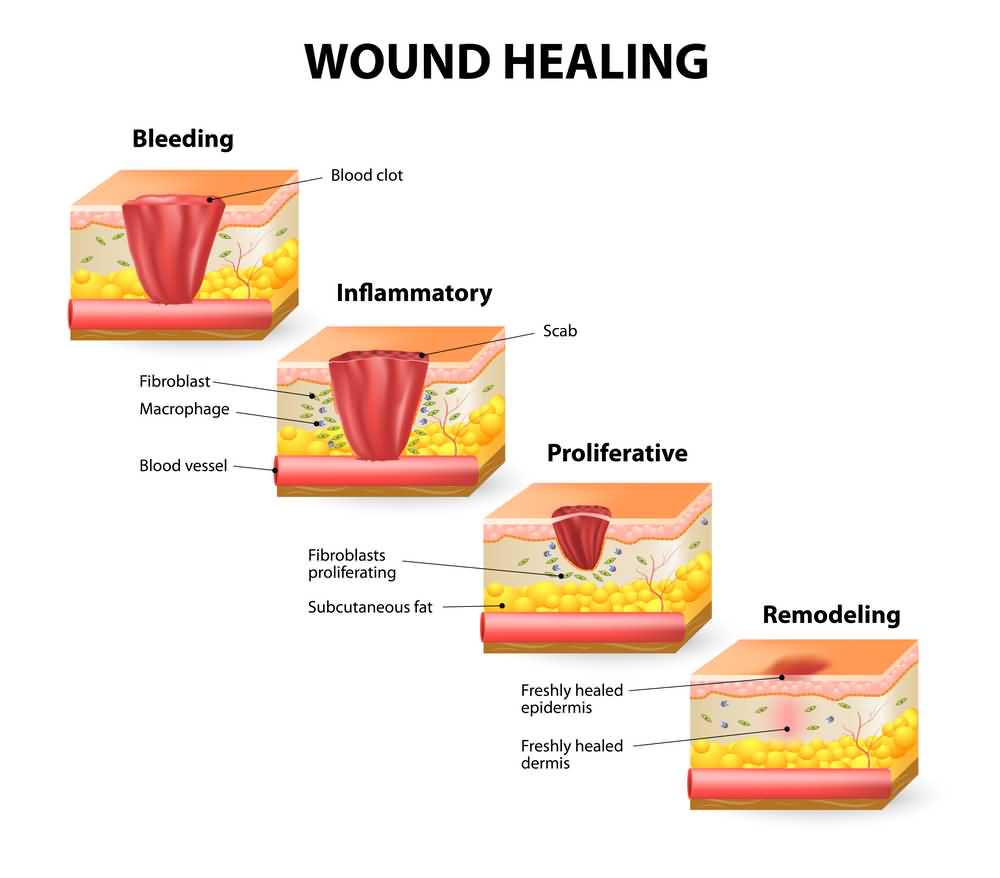

This is one of the most common manifestations of hemophilia regardless of its severity. The clotting factors contribute to healing wounds by creating a blood clot that will then be covered with healthy tissue. In hemophilia, this does not happen as it should, and patients continue bleeding without control. Thus, doctors are cautious with these individuals if they ever need surgery, and special measures are taken if that’s the case.
2. Abdominal organ bleeding


Without any notice, and in severe cases of hemophilia, patients can have severe internal bleeding episodes. One of the abdominal organs that usually bleed is the spleen, usually after trauma. But in patients with severe hemophilia, the incidence of abdominal bleeding is higher and very dangerous. In extreme cases, it causes severe anemia, and sometimes death.
3. Bleeding in the joints


The joints are subject to continuous wear and tear. Thus, bleeding in the joints is a common problem in patients with hemophilia. We shouldn’t have blood in the joints, so this causes tightness and joint pain. The joint starts swelling and becomes tender. The most commonly affected joints are those of the knees, ankles, and elbows.
4. Bleeding inside the skull
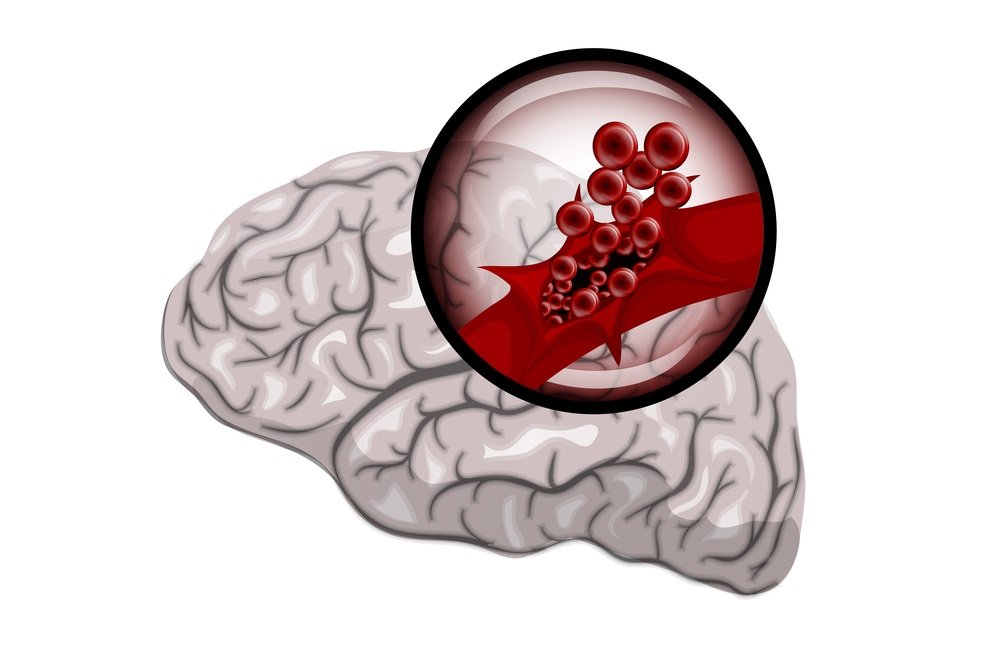

Bleeding in the brain structures is very dangerous, and individuals with hemophilia are susceptible to hemorrhagic stroke, seizures, paralysis, and other associated problems. Bleeding inside the head may also result from delivery when doctors need to use forceps and other aggressive methods to facilitate delivery.
5. Bruises and skin manifestations


This is also a ubiquitous symptom, even in mild and moderate hemophilia cases. These patients have bleeding in the skin, which manifests as bruises, petechiae, and other skin manifestations. The blood builds up in the soft tissue, and we can also have large hematomas in the affected area. These skin manifestations are usually followed by mild trauma or after pressing on the skin, hard enough to cause microscopic lesions that won’t be healed and continue bleeding indeterminately.
6. Gum and mouth bleeding

This is also a common symptom in patients with mild and moderate hemophilia cases. Gum bleeding is usually evident after brushing our teeth or eating hard foods. Bleeding in the mouth may also be associated with foul breath and may become a dangerous hemorrhage after the patient loses a tooth or after tooth extraction.
7. Continuous bleeding after injections and vaccines


We all may experience minor bleeding after a shot. But patients with hemophilia continue bleeding from the same spot, regardless of the attempts to press on the area or using an adhesive bandage.
8. Bleeding in the urine

It is not unusual to find bleeding in the urine if you have hemophilia. Naturally, this does not mean that you should take it for granted. It can also be a symptom of urinary tract infections, kidney stones, and other urologic problems.
9. Nosebleeds

The blood vessels in the nostrils are tiny and fragile. They can be broken or injured after a forceful nose blowing or sneezing. The same happens in certain weather conditions or when blood pressure is high. In patients with hemophilia, this nose bleeding can be severe and very difficult to manage, and they do have more episodes compared to the average patient.
10. Gastrointestinal bleeding

Another common site of bleeding comes from the digestive tube. It can be red bleeding, usually from the rectum or anus, especially in constipated patients. Or it may be dark-colored and with a foul smell, which is known as melena and comes from bleeding in the upper gastrointestinal tract.
11. Coughing up or vomiting blood


Both conditions should be carefully evaluated. The former is known as hemoptysis and comes from bleeding in the respiratory tract. The latter is known as hematemesis, and it comes from the upper gastrointestinal tract.
12. General symptoms

Secondary to bleeding, we can also have general symptoms, most of them associated with anemia. They include weakness, tachycardia (rapid heartbeats), tachypnea (fast breathing frequency), and orthostasis (feeling dizzy after changing posture).
13. Tingling, stiffness, and refusal to use the affected joints

In some patients with joint involvement, they do not have severe pain and swelling but the tingling and mild stiffness. It is also important to note that children may refuse to use the affected joint and become irritable when forced to move them.
Diagnosis


Since hemophilia runs in families and is more frequent in males, diagnosis is usually made early in life. After birth, those with family members with hemophilia typically ask for a test to ensure that their baby boys do not have the disease.
The diagnosis starts by performing a few blood tests to evaluate the clotting function. If clotting tests are not normal, they proceed to use more specific tests to assess the disorder’s cause. They are clotting factor tests and help doctors to locate which clotting factor is absent or abnormal, thus diagnosing the type of hemophilia.
The workup should include the following tests:
- Complete blood count: In this test, it is essential to measure hemoglobin levels, the number of red blood cells, and their size. In most cases, CBC is normal, but sometimes it may show anemia, especially after uncontrolled bleeding episodes.
- Activated Partial Thromboplastin Time Test: This test evaluates the clotting ability of several factors in the blood (FVIII, FIX, FXI, and FXII). Having a deficient level of these clotting factors is the first step to diagnosing hemophilia A or hemophilia B. Since this test does not differentiate FVIII from FIX abnormalities, other tests must make the distinction.
- Prothrombin Time Test: This test evaluates the clotting ability of several factors in the blood (FI, FII, FV, FVII, FX). These tests are usually normal in patients with hemophilia, but the test should be taken to rule out other diseases.
- Fibrinogen Test: This test evaluates the clotting factor I by itself and helps doctors evaluate the patient if he has an abnormal result in any of the tests above.
- Clotting factor tests: In patients with an abnormal result in clotting tests, a clotting factor test is then performed to precisely determine which clotting factor is causing their bleeding disorder. This test is important to differentiate hemophilia A from hemophilia B. It is also an accurate measure of the severity of the condition. When FVIII and FIX are 50-100%, the individual is normal. We can diagnose mild hemophilia when results are lower than 50%. Moderate hemophilia is diagnosed when FVIII or FIX levels are shorter than 5%. And severe hemophilia is diagnosed with clotting factor levels lower than 1%.
- Testing for inhibitors: This exam is usually performed in moderate and severe hemophilia cases and patients who do not have an appropriate response to treatment. It is commonly measured using the Bethesda method, with a positive result whenever we have 0.6 Bethesda Units (BU) or more. 5 BU or more is associated with more severe disease.
- Radiography: In patients with hemophilia, one of the most common manifestations is associated with joint health. Thus, radiography is important to evaluate the joints. Acute hemarthrosis is difficult to consider, but we can assess chronic degenerative joint disease with radiographs. It is essential to assess cartilage damage, bone cyst formation, synovial hypertrophy, and other joint problems commonly found in hemophilia.
Treatment & therapy


Hemophilia treatment is usually done by replacing or administering the clotting factor that is abnormal or missing. This is a protein we can infuse in the blood through a vein, and there are commercial concentrates we can use for this purpose. After some time, patients with hemophilia learn to use these commercial concentrates by themselves whenever they have a bleeding episode.
Besides using the clotting factor concentrates to stop bleeding, we can also use them as a prophylaxis to prevent new bleeding episodes. By doing this, it is possible to avoid severe bleeding episodes, especially before specific medical procedures.
Treatment is more difficult and expensive when hemophilia is caused by an inhibitor that prevents clotting factors from functioning correctly. These patients need more clotting factors than usual or another type of clotting factor that is even more expensive.
Treatment is usually administered at special medical facilities known as Hemophilia Treatment Centers. In these facilities, professionals will address medical treatment and provide the education and instruction hemophilia patients need to improve their quality of life.
Treatment medications typically include:
- Plasma-derived factor concentrates: It’s a concentrate of plasma, which is the yellow-colored liquid of the blood. It contains several proteins, including albumin and all of the clotting factors. Plasma is tested to make sure they do not carry any virus, and then it is separated into concentrates of its components. In this case, we use a concentrate of clotting factors, which are freeze-dried to make sure they do not contain pathogens.
- Recombinant factor concentrates: It is another alternative that does not come from human plasma. Instead, recombinant factor concentrates are genetically engineered and prepared for commercial use. They do not have any plasma particle, albumin, or anything else that may potentially carry a virus. The downside of using a recombinant factor concentrate is that it brings a higher chance of developing inhibitors.
Other therapeutic options available for patients with hemophilia include:
- Emicizumab: The commercial name is Hemlibra, and it is also known as ACE 910. It replaces the function of FVIII and can be used to prevent bleeding episodes in hemophilia A. It is injected under the skin, but similar to recombinant factor concentrates, this therapy carries a higher chance of developing inhibitors.
- Desmopressin Acetate: The commercial name is Stimate or DDAVP. This drug works similarly to a hormone in the body that releases more FVIII. It is appropriate in cases of mild hemophilia and applied intravenously or through a nasal spray.
- Epsilon Amino Caproic Acid: The commercial name is Amicar and has the ability to prevent the breakdown of blood clots. Thus, patients will have a firmer and more resistant clot that promotes hemostasis. It is commonly used in order to avoid gum or mouth bleeding, which is inhibited by substances in the saliva.
Prognosis & life expectancy


With appropriate treatment, the prognosis of patients with hemophilia is very favorable. They can live productive lives, especially when they are treated early and use prophylaxis measures.
Still, a small proportion of patients with hemophilia develop a very severe type. One-quarter of them end up with impaired motor skills, behavioral and cognitive problems by their 6-18 years of age.
Life expectancy in cases of hemophilia is 60 years or more.
Hemophilia in women


Women can have hemophilia, too, but it is not as common as in their male counterparts. Women have two X chromosomes, and hemophilia is a recessive disorder, which means that the disease should take both X chromosomes before they manifest any symptom. A woman with hemophilia is likely born from a mother and father with hemophilia or a father with hemophilia and a carrier mother.
References
Hartmann, J., & Croteau, S. E. (2016). 2017 Clinical trials update: Innovations in hemophilia therapy. American journal of hematology, 91(12), 1252-1260.
Kruse‐Jarres, R., Kempton, C. L., Baudo, F., Collins, P. W., Knoebl, P., Leissinger, C. A., … & Kessler, C. M. (2017). Acquired hemophilia A: updated review of evidence and treatment guidance. American Journal of Hematology, 92(7), 695-705.
Bertamino, M., Riccardi, F., Banov, L., Svahn, J., & Molinari, A. C. (2017). Hemophilia care in the pediatric age. Journal of clinical medicine, 6(5), 54.
Ljung, R. (2016). Aspects of prophylactic treatment of hemophilia. Thrombosis journal, 14(1), 30.